Summary
Clinical characteristics.
Hypermanganesemia with dystonia 1 (HMNDYT1) is characterized by the following:
- A movement disorder resulting from manganese accumulation in the basal ganglia
- Whole-blood manganese concentrations that often exceed 2000 nmol/L (normal: <320 nmol/L)
- Polycythemia
- Hepatomegaly with variable hepatic fibrosis/cirrhosis
Neurologic findings can manifest in childhood (ages 2-15 years) as four-limb dystonia, leading to a characteristic high-stepping gait ("cock-walk gait"), dysarthria, fine tremor, and bradykinesia or on occasion spastic paraplegia; or in adulthood as parkinsonism (shuffling gait, rigidity, bradykinesia, hypomimia, and monotone speech) unresponsive to L-dopa treatment.
Diagnosis/testing.
The diagnosis of HMNDYT1 is established in a proband with suggestive findings and biallelic pathogenic variants in SLC30A10 identified by molecular genetic testing.
Management.
Treatment of manifestations: Regular chelation therapy with intravenous disodium calcium edetate improves blood manganese levels and neurologic findings and halts liver disease. In addition, supplementation with oral iron therapy (despite normal serum iron levels) can reduce blood manganese levels and resolve polycythemia. The potential for complications from chelation therapy and/or iron supplementation can be lessened by careful monitoring. Physical therapy (to prevent contractures and maintain ambulation), occupational therapy, and/or speech therapy and use of adaptive aids and assistive communication devices are recommended. Progressive dystonia may necessitate a gastrostomy tube for adequate nutrition and a tracheostomy may be needed to prevent aspiration pneumonia.
Prevention of primary manifestations: Chelation therapy and iron supplementation may prevent primary disease manifestations in affected asymptomatic sibs.
Agents/circumstances to avoid: Foods very high in manganese: cloves; saffron; nuts; mussels; dark chocolate; and pumpkin, sesame, and sunflower seeds.
Evaluation of relatives at risk: Because chelation therapy and iron supplementation could prevent primary disease manifestations in affected asymptomatic individuals, it is recommended that at-risk sibs of a proband be evaluated either by molecular genetic testing (if the pathogenic variants in the family are known) or by periodic monitoring of whole-blood manganese concentration and hemoglobin.
Genetic counseling.
HMNDYT1 is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an SLC30A10 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial pathogenic variants. Once the SLC30A10 pathogenic variants have been identified in an affected family member, carrier testing for at-risk family members and prenatal and preimplantation genetic testing are possible.
Diagnosis
Hypermanganesemia with dystonia 1 (HMNDYT1) presents as a movement disorder associated with manganese accumulation in the basal ganglia. No consensus clinical diagnostic criteria have been published.
Suggestive Findings
HMNDYT1 should be suspected in individuals with typical clinical, brain MRI, and laboratory findings and family history.
Clinical Findings
An early- and a late-onset form exist:
- Childhood-onset form (between ages 2 and 15 years). Usually four-limb dystonia, leading to a characteristic high-stepping gait ("cock-walk gait"), dysarthria, fine tremor, and bradykinesia [Tuschl et al 2012, Quadri et al 2015] or on occasion spastic paraplegia [Gospe et al 2000]
- Adult-onset form. Parkinsonism (shuffling gait, rigidity, bradykinesia, hypomimia, and monotone speech) unresponsive to L-dopa treatment [Quadri et al 2012]
Brain MRI
T1-weighted images show characteristic hyperintensity of the basal ganglia including the globus pallidus; putamen; and caudate, subthalamic, and dentate nuclei with sparing of the thalamus and ventral pons. When the disease is extensive, white matter and anterior pituitary involvement can be present (Figure 1).
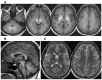
Figure 1.
Representative brain MRI of an affected individual A. Transaxial T1-weighted images. Note abnormally high signal return from all white matter as well as more prominent signal return from the putamen and globus pallidus bilaterally.
T2-weighted images show corresponding hypointensity changes. However, these changes are often less pronounced and, hence, may be reported as normal (see Figure 1).
Note: Normalization of manganese blood levels (see Management) improves findings on brain MRI [Quadri et al 2012, Stamelou et al 2012, Tuschl et al 2012].
Laboratory Findings
Hypermanganesemia
- Whole-blood manganese concentrations are elevated in all affected individuals. Average in affected individuals is greater than 2,000 nmol/L (normal: <320 nmol/L).
- In contrast, blood manganese concentration in acquired hypermanganesemia is usually less than 2,000 nmol/L.
Corroborative laboratory features
- Polycythemia. Manganese induces expression of the gene encoding erythropoietin [Ebert & Bunn 1999]. Characteristically, affected individuals are polycythemic. Hemoglobin concentrations reported in the literature range from 15.9 to 22.5 g/dL (mean: 18.6 g/dL). Some individuals studied have had elevated erythropoietin levels [Gospe et al 2000, Quadri et al 2012, Tuschl et al 2012].
- Markers of depleted iron stores. Manganese and iron compete for the same serum-binding protein (transferrin) and membranous transporter protein (divalent metal transporter 1). Therefore, affected individuals show low serum ferritin concentration and serum iron levels while total iron-binding capacity is elevated [Quadri et al 2012, Tuschl et al 2012].
- Chronic liver disease. Hepatic involvement may be present with variable severity and is not pathognomonic for this disease; when present, however, hepatic involvement should further suggest the diagnosis:
- The majority of affected individuals reported to date have evidence of hepatic involvement that includes hepatomegaly, elevated transaminases (alanine transaminase, aspartate transaminase), and unconjugated hyperbilirubinemia.
- Liver ultrasound examination or MRI can confirm hepatomegaly and features of liver cirrhosis.
- Pathologic features on liver biopsy / postmortem examination in six affected individuals included fibrosis, steatosis, and micronodular cirrhosis.
- Note: One individual with hepatomegaly and micronodular cirrhosis had no laboratory evidence of hepatic dysfunction [Gospe et al 2000, Tuschl et al 2008, Quadri et al 2012, Tuschl et al 2012, Lechpammer et al 2014].
- Hepatic manganese content is highly elevated. Rhodanine staining confirms deposition of manganese in hepatocytes. Copper and zinc content can also be affected with mild elevation in hepatic levels [Gospe et al 2000, Tuschl et al 2008, Quadri et al 2012, Tuschl et al 2012, Lechpammer et al 2014].
Family History
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of HMNDYT1 is established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in SLC30A10 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic SLC30A10 variants of uncertain significance (or of one known SLC30A10 pathogenic variant and one SLC30A10 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with atypical findings or in whom the diagnosis of HMNDYT1 has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SLC30A10 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A multigene panel that includes SLC30A10 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hypermanganesemia with Dystonia 1
Clinical Characteristics
Clinical Description
To date, 48 individuals have been identified with biallelic pathogenic variants in SLC30A10 [Anagianni & Tuschl 2019, Tavasoli et al 2019, Yapici et al 2019, Lambrianides et al 2020]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Hypermanganesemia with Dystonia 1: Frequency of Select Features
Neurologic Findings
Childhood onset. In the childhood-onset form of hypermanganesemia with dystonia 1 (HMNDYT1), affected individuals present with neurologic signs between ages two and 15 years. Many become wheelchair bound in their teens.
The neurologic signs and symptoms of the childhood-onset form are primarily extrapyramidal and include dystonia, dysarthria, and rigidity. Four-limb dystonia manifests with difficulties walking and a high-stepping gait ("cock walk gait"), dystonic posturing, and painful extensor spasms. Fine motor impairment causes problems with writing and drawing and inability to perform rapid alternating movements of the hands (dysdiadochokinesis). Dystonia of the tongue can lead to dysarthria [Quadri et al 2012, Tuschl et al 2012, Quadri et al 2015]. Dystonia may lead to progressive swallowing difficulties and aspiration which may require gastrostomy/tracheostomy insertion.
Isolated corticospinal tract involvement has been described in one affected individual. Typical neurologic signs of spastic paraparesis (e.g., spasticity, hyperreflexia, extensor plantar responses) were found [Gospe et al 2000].
Adult onset. Quadri et al [2012] reported two brothers who presented at ages 47 years and 57 years with progressive gait disturbance and bradykinesia. Neurologic examination showed features of parkinsonism including hypomimia, monotone speech, mild rigidity, global bradykinesia, wide-based gait with freezing and starting hesitation, and moderate postural instability without evidence of tremor, dystonia, or cerebellar or pyramidal disturbances. Treatment with L-dopa and dopamine agonists did not improve neurologic findings.
Sensorimotor axonal polyneuropathy has been described in two affected individuals with the late-onset neurologic presentation [Quadri et al 2012].
Hypermanganesemia
Whole-blood manganese concentrations are elevated in the majority of affected individuals.
Due to limited data, the onset of hypermanganesemia is not accurately known. Raised whole-blood manganese concentrations have been recorded in affected children as young as age three years [Quadri et al 2015]; however, given that clinical manifestations can be apparent in the first two years of life, it is expected that hypermanganesemia develops concurrently with or prior to onset of clinical manifestations.
Hypermanganesemia due to environmental overexposure (including parenteral nutrition) and acquired hepatocerebral degeneration in persons with end-stage liver disease must be excluded. See Differential Diagnosis.
Quadri et al [2012] reported one affected individual whose blood manganese concentration was only minimally increased on one occasion; similarly, Lambrianides et al [2020] reported an affected individual with evidence of manganese deposition on brain MRI but normal blood manganese levels in adulthood. Therefore, normal blood manganese levels do not exclude a diagnosis of HMNDYT1 when the clinical suspicion is strong.
Note: Blood manganese concentrations of heterozygotes (i.e., carriers of one SLC30A10 pathogenic variant) are within normal limits or are mildly elevated. Gospe et al [2000] reported a borderline high blood manganese concentration of 380 nmol/L in an obligate heterozygous parent and Tuschl et al [2012] reported levels between 380 and 649 nmol/L in three heterozygous parents (normal: <320 nmol/L).
Polycythemia
All affected individuals reported to date had polycythemia at the time of diagnosis. Polycythemia can precede the onset of neurologic manifestations; therefore, affected individuals often undergo repeat phlebotomies prior to identification of the correct diagnosis [Quadri et al 2012, Tuschl et al 2012]. Polycythemia has been described in affected children from age three years; earlier presentation of polycythemia cannot be ruled out due to insufficient data. Individuals in whom neurologic symptoms do not manifest until late adulthood have had polycythemia since as early as the third decade. Polycythemia can resolve on treatment with chelation therapy or iron. There is evidence from one patient whose polycythemia resolved without treatment during advanced stage of disease [Lechpammer et al 2014].
Liver Disease
The spectrum of hepatic involvement ranges from mild hepatomegaly to hepatic failure in early adulthood [Tuschl et al 2012]. However, pure neurologic phenotypes presenting with dystonia alone have also been reported [Quadri et al 2012].
In the majority of affected individuals, transaminases are mildly elevated [Quadri et al 2012, Tuschl et al 2012]. To date, three affected individuals died of complications of liver cirrhosis between ages 18 and 46 years. As most of the affected individuals known to the authors are still in their teens or early adulthood, no long-term follow-up data are available.
Significant phenotypic variability with regard to hepatic involvement is apparent even within the same family: The two brothers reported by Quadri et al [2012], who are now in their sixties and severely affected by dystonia, did not show hepatic involvement. Both had normal liver function and liver ultrasound examination throughout their lives. However, while the affected sister had minimal neurologic involvement, she developed liver cirrhosis in the third decade and died of liver failure at age 46 years.
Other
Intellect appears normal in all affected individuals. Quadri et al [2012] described one individual who developed cognitive and behavioral problems, thought to be alcohol related. While environmental manganese exposure is known to cause cognitive and psychiatric disturbances ("manganese madness") including emotional lability, hallucinations, and compulsive behavior [Racette et al 2012], this has not yet been observed in individuals with hypermanganesemia with dystonia 1.
Pica. Several affected individuals had pica during early childhood [Brna et al 2011; Brna, unpublished data].
Darker skin tone. Some affected individuals have been described to have a purple or dark skin discoloration to an extent that parents are able to distinguish affected and unaffected children prior to the manifestation of clinical symptoms [Authors, unpublished data].
Pathology
Postmortem studies in an individual with SLC30A10 deficiency showed yellow-gray mottling of the basal ganglia associated with severe neuronal loss, astrocytosis, myelin loss, spongiosis, and rhodanine-positive deposits particularly in the globus pallidus, while other basal ganglia were affected to a lesser extent. Gliosis of the white matter and axonal loss of the corticospinal tracts were observed [Lechpammer et al 2014].
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Prevalence
A total of 48 affected individuals from 25 families are known worldwide [Quadri et al 2012, Tuschl et al 2012, Avelino et al 2014, Quadri et al 2015, Mukhtiar et al 2016, Anagianni & Tuschl 2019, Tavasoli et al 2019, Yapici et al 2019, Lambrianides et al 2020]. The prevalence is yet to be determined.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants SLC30A10.
Differential Diagnosis
Acquired hypermanganesemia. Overexposure to manganese is known to be neurotoxic and causes "manganism" – a distinct syndrome of extrapyramidal movement disorder (dystonia/parkinsonism) combined with high signal intensity of the basal ganglia on T1-weighted MR images of the brain resulting from manganese accumulation in the basal ganglia [Racette et al 2012].
- Environmental exposure has been described in workers in mining and welding industries who inhale manganese-laden dust or fumes, in individuals ingesting contaminated drinking water, and in drug addicts who use intravenous methcathinone contaminated with potassium permanganate [Bouchard et al 2011, Racette et al 2012, Ordak et al 2022].
- Total parenteral nutrition has been associated with manganese toxicity because the control mechanisms of manganese absorption in the gut and subsequent hepatic excretion are bypassed [Chalela et al 2011].
- Acquired hepatocerebral degeneration is observed in those with advanced hepatic cirrhosis or portosystemic shunts, in which impaired biliary excretion of manganese results in manganese accumulation in the basal ganglia, causing a debilitating movement disorder [Meissner & Tison 2011].
Other conditions to consider in the differential diagnosis of hypermanganesemia with dystonia 1 (HMNDYT1) include the following:
- SLC39A14 deficiency (SLC39A14-related early-onset parkinsonism-dystonia, hypermanganesemia with dystonia 2 [HMNDYT2]): a manganese transporter defect caused by impaired manganese uptake into the liver and gut;
- Parkinson disease and its differential diagnoses (atypical degenerative parkinsonisms [multiple-system atrophy, progressive supranucleal palsy], vascular parkinsonism, and drug-induced parkinsonism)
- Hereditary dystonia
- Neurodegenerative diseases associated with dystonia (including organic acidemias [i.e. glutaric, methylmalonic, propionic, 3-hydroxyisobutyryl-CoA hydrolase deficiency])
- Cerebral palsy
Table 3 lists associated genes and clinical characteristics of SLC39A14 deficiency and Wilson disease and provides selected examples of genes known to be involved in monogenic Parkinson disease, hereditary dystonia, neurodegenerative diseases associated with dystonia, and hereditary spastic paraplegia (which can resemble spastic diplegic cerebral palsy in individuals with onset in early childhood).
Table 3.
Hereditary Disorders in the Differential Diagnosis of Hypermanganesemia with Dystonia 1
Management
No clinical practice guidelines for hypermanganesemia with dystonia 1 (HMNDYT1) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with HMNDYT1, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Hypermanganesemia with Dystonia 1
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with Hypermanganesemia with Dystonia 1
Adverse Effects of Chelation Therapy
Adverse effects of chelation therapy with disodium calcium edetate include hypocalcemia, nephrotoxicity, trace metal and vitamin deficiency, and thrombocytopenia and leukopenia [Lamas et al 2012].
Treatment may need to be discontinued if the following occur:
- White blood count <3.5x109/L
- Neutrophils <2x109/L
- Platelets <150x109/L
- >2+ proteinuria on >1 occasion (and no evidence of infection)
The above cut-off values are based on guidelines for D-penicillamine treatment [Chakravarty et al 2008]. The clinical treatment benefit needs to be carefully weighed against occurring adverse effects for each affected individual.
Prevention of Primary Manifestations
Chelation therapy and iron supplementation may prevent primary disease manifestations in affected sibs who are asymptomatic (see Treatment of Manifestations).
Surveillance
Table 6.
Recommended Surveillance for Individuals with Hypermanganesemia with Dystonia 1
Agents/Circumstances to Avoid
Foods very high in manganese (cloves; saffron; nuts; mussels; dark chocolate; pumpkin, sesame, and sunflower seeds) should be avoided.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic sibs of a proband in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures. Chelation therapy and iron supplementation can potentially prevent primary disease manifestations in affected sibs who are asymptomatic (see Treatment of Manifestations).
Periodic monitoring of whole-blood manganese concentration and hemoglobin is recommended if the genetic status of a sib is unknown (i.e., if a sib has not undergone molecular genetic testing for the SLC30A10 pathogenic variants identified in the proband).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
For an affected fetus, no prenatal treatment is recommended as the disease does not manifest before early childhood.
For an affected mother, no data or information on pregnancy management are available.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Hypermanganesemia with dystonia 1 (HMNDYT1) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., presumed to be carriers of one SLC30A10 pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an SLC30A10 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an SLC30A10 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial pathogenic variants.
- Significant phenotypic variability may be observed between affected sibs, particularly with regard to liver disease, which may be absent or mild in some individuals while their sibs develop chronic liver disease and associated complications.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband
- No data on fertility in individuals with HMNDYT1 are available.
- Assuming that reproduction is possible, the offspring of an affected individual are obligate heterozygotes (carriers) for a pathogenic variant in SLC30A10.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an SLC30A10 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SLC30A10 pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC30A10 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for a pregnancy at increased risk are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- American Liver FoundationPhone: 800-465-4837 (HelpLine)
- Dystonia Medical Research FoundationPhone: 312-755-0198; 800-377-DYST (3978)Fax: 312-803-0138Email: dystonia@dystonia-foundation.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hypermanganesemia with Dystonia 1: Genes and Databases
Table B.
OMIM Entries for Hypermanganesemia with Dystonia 1 (View All in OMIM)
Molecular Pathogenesis
SLC30A10 is a member of the SLC30 solute carrier subfamily of the cation diffusion facilitator (CDF) family. Human SLC30A10 is a protein of 485 amino acids [Tuschl et al 2012]. The protein is a transmembrane manganese transporter expressed in liver and brain that facilitates manganese efflux at the cell surface [Leyva-Illades et al 2014]. Quadri et al [2012] showed that SLC30A10 expression and the levels of the encoded protein are under strict control by extracellular manganese levels in vitro. Exposure to high manganese concentrations leads to significant increase of SLC30A10 mRNA and protein expression.
Pathogenic variants in SLC30A10 have deleterious effects on protein function. While wild type SLC30A10 expressed in manganese-sensitive yeast cells rescues growth in high manganese concentrations, SLC30A10 with missense and nonsense sequence changes fails to restore manganese resistance [Tuschl et al 2012]. Furthermore, SLC30A10 pathogenic variants lead to loss of immunoreactivity in liver and brain tissues of affected individuals and fail to traffic to the cell surface [Quadri et al 2012, Lechpammer et al 2014, Leyva-Illades et al 2014]. In humans, impaired function of SLC30A10 results in accumulation of manganese in liver and brain [Gospe et al 2000, Lechpammer et al 2014].
Mechanism of disease causation. Hypermanganesemia with dystonia 1 (HMNDYT1) occurs by a loss-of-function mechanism. Pathogenic variants are predicted to either (1) cause a significantly truncated protein because of a frameshift and premature stop codon or large deletion, or (2) affect an evolutionary highly conserved area of the protein. Therefore, these sequence changes have detrimental effects on protein function [Quadri et al 2012, Tuschl et al 2012].
SLC30A10 localizes to the apical domain of hepatocytes and enterocytes where it facilitates manganese excretion. Studies in mice have confirmed that loss of SLC30A10 function leads to impaired biliary and intestinal manganese elimination with subsequent accumulation of manganese in the liver and brain [Mercadante et al 2019, Taylor et al 2019]. Furthermore, SLC30A10 expressed in the brain protects from any increase in manganese and neurotoxicity [Taylor et al 2019].
Chapter Notes
Author Notes
The authors are studying the mechanisms underlying manganese neurotoxicity and inherited manganese transporter defects at the University College London (UCL) Great Ormond Street Institute of Child Health.
Tuschl Lab website: zebrafishucl.org/tuschl
Dr Karin Tuschl UCL website: www.ucl.ac.uk
Revision History
- 23 December 2021 (ha) Comprehensive update posted live
- 9 February 2017 (ha) Comprehensive update posted live
- 11 September 2014 (me) Comprehensive update posted live
- 30 August 2012 (me) Review posted live
- 1 June 2012 (kt) Original submission
References
Literature Cited
- Anagianni S, Tuschl K. Genetic disorders of manganese metabolism. Curr Neurol Neurosci Rep. 2019;19:33. [PMC free article: PMC6517356] [PubMed: 31089831]
- Avelino MA, Fusão EF, Pedroso JL, Arita JH, Ribeiro RT, Pinho RS, Tuschl K, Barsottini OG, Masruha MR. Inherited manganism: the "cock-walk" gait and typical neuroimaging features. J Neurol Sci. 2014;341:150–2. [PubMed: 24746291]
- Bouchard MF, Sauvé S, Barbeau B, Legrand M, Brodeur MÈ, Bouffard T, Limoges E, Bellinger DC, Mergler D. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect. 2011;119:138–43. [PMC free article: PMC3018493] [PubMed: 20855239]
- Brna P, Gordon K, Dooley JM, Price V. Manganese toxicity in a child with iron deficiency and polycythemia. J Child Neurol. 2011;26:891–4. [PubMed: 21596707]
- Chakravarty K, McDonald H, Pullar T, Taggart A, Chalmers R, Oliver S, Mooney J, Somerville M, Bosworth A, Kennedy T. BSR/BHPR guideline for disease-modifying anti-rheumatic drug (DMARD) therapy in consultation with the British Association of Dermatologists. Rheumatology. 2008;47:924–5. [PubMed: 16940305]
- Chalela JA, Bonillha L, Neyens R, Hays A. Manganese encephalopathy: an under-recognized condition in the intensive care unit. Neurocrit Care. 2011;14:456–8. [PubMed: 21174173]
- Ebert BL, Bunn HF. Regulation of the erythropoietin gene. Blood. 1999;94:1864–77. [PubMed: 10477715]
- Gospe SM Jr, Caruso RD, Clegg MS, Keen CL, Pimstone NR, Ducore JM, Gettner SS, Kreutzer RA. Paraparesis, hypermanganesaemia, and polycythaemia: a novel presentation of cirrhosis. Arch Dis Child. 2000;83:439–42. [PMC free article: PMC1718535] [PubMed: 11040156]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Lamas GA, Goertz C, Boineau R, Mark DB, Rozema T, Nahin RL, Drisko JA, Lee KL. Design of the Trial to Assess Chelation Therapy (TACT). Am Heart J. 2012;163:7–12. [PMC free article: PMC3243954] [PubMed: 22172430]
- Lambrianides S, Nicolaou P, Michaelidou M, Kakouris P, Votsi C, Petrou PP, Drousiotou A, Minaidou A, Demetriou P, Voulgaris C, Christodoulou K, Tanteles GA, Pantzaris M. A novel SLC30A10 missense variant associated with parkinsonism and dystonia without hypermanganesemia. J Neurol Sci. 2020;418:117101. [PubMed: 32866815]
- Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM Jr. Pathology of inherited manganese transporter deficiency. Ann Neurol. 2014;75:608–12. [PubMed: 24599576]
- Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. J Neurosci. 2014;34:14079–95. [PMC free article: PMC4198546] [PubMed: 25319704]
- Meissner W, Tison F. Acquired hepatocerebral degeneration. Handb Clin Neurol. 2011;100:193–7. [PubMed: 21496578]
- Mercadante CJ, Prajapati M, Conboy HL, Dash ME, Herrera C, Pettiglio MA, Cintron-Rivera L, Salesky MA, Rao DB, Bartnikas TB. Manganese transporter Slc30a10 controls physiological manganese excretion and toxicity. J Clin Invest. 2019;129:5442–5461. [PMC free article: PMC6877324] [PubMed: 31527311]
- Mukhtiar K, Ibrahim S, Tuschl K, Mills P. Hypermanganesemia with dystonia, polycythemia and cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain Dev. 2016;38:862–5. [PubMed: 27117033]
- Ordak M, Sloniewicz N, Nasierowski T, Muszynska E, Bujalska-Zadrozny M. Manganese concentration in patients with encephalopathy following ephedrone use: a narrative review and analysis of case reports. Clin Toxicol (Phila). 2022;60:10-17. [PubMed: 34521308]
- Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, Severijnen LA, Di Toro Mammarella L, Mignarri A, Monti L, Sanna A, Lu P, Punzo F, Cossu G, Willemsen R, Rasi F, Oostra BA, van de Warrenburg BP, Bonifati V. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90:467–77. [PMC free article: PMC3309204] [PubMed: 22341971]
- Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, Kori I, Hattiholi V, Jain P, Aneja S, Kumar A, Gulati P, Goel M, Talukdar B, Bonifati V. Manganese transport disorder: novel SLC30A10 mutations and early phenotypes. Mov Disord. 2015;30:996–1001. [PubMed: 25778823]
- Racette BA, Aschner M, Guilarte TR, Dydak U, Criswell SR, Zheng W. Pathophysiology of manganese-associated neurotoxicity. Neurotoxicology. 2012;33:881–6. [PMC free article: PMC3350837] [PubMed: 22202748]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Stamelou M, Tuschl K, Chong WK, Burroughs AK, Mills PB, Bhatia KP, Clayton PT. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov Disord. 2012;27:1317–22. [PMC free article: PMC3664426] [PubMed: 22926781]
- Tavasoli A, Rafsanjani KA, Hemmati S, Mojbafan M, Zarei E, Hosseini S. A case of dystonia with polycythemia and hypermanganesemia caused by SLC30A10 mutation: a treatable inborn error of manganese metabolism. BMC Pediatr. 2019;19:229. [PMC free article: PMC6615235] [PubMed: 31288771]
- Taylor CA, Hutchens S, Liu C, Jursa T, Shawlot W, Aschner M, Smith DR, Mukhopadhyay S. SLC30A10 transporter in the digestive system regulates brain manganese under basal conditions while brain SLC30A10 protects against neurotoxicity. J Biol Chem. 2019;294:1860–76. [PMC free article: PMC6369308] [PubMed: 30559290]
- Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, Aulakh R, Ribeiro RT, Barsottini OG, Zaki MS, Del Rosario ML, Dyack S, Price V, Rideout A, Gordon K, Wevers RA, Kling Chong WK, Mills PB. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90:457–66. [PMC free article: PMC3309187] [PubMed: 22341972]
- Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, Clayton PT. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia-A new metabolic disorder. J Inherit Metab Dis. 2008;31:151–63. [PubMed: 18392750]
- Yapici Z, Tuschl K, Eraksoy M. Hypermanganesemia with dystonia 1: a novel mutation and response to iron supplementation. Mov Disord Clin Pract. 2019;7:94–96. [PMC free article: PMC6962664] [PubMed: 31970220]
Publication Details
Author Information and Affiliations
UCL Great Ormond Street Institute of Child Health
London, United Kingdom
UCL Great Ormond Street Institute of Child Health
London, United Kingdom
University of Washington
Seattle, Washington
UCL Great Ormond Street Institute of Child Health
London, United Kingdom
Publication History
Initial Posting: August 30, 2012; Last Update: December 23, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Tuschl K, Clayton PT, Gospe SM Jr, et al. Hypermanganesemia with Dystonia 1. 2012 Aug 30 [Updated 2021 Dec 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
