

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
NCBI News [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 1991-2012.

Specialized database with unique search interface added to Zika virus resource page
Thursday, March 31, 2016
The NCBI Zika virus resource page has been updated with a specialized database. This database uses pipelines to annotate genes, proteins and mature peptides, and standardize sample metadata. With this database, you can:
- Find sequences easily using standardized annotations and normalized metadata terms
- Construct alignments and phylogenetic trees using a suite of online tools
- Download sequences and metadata in a variety of formats and create customized titles/deflines for FASTA file downloads.
The NCBI Zika virus resource, part of the Virus Variation family of NCBI resources, provides users with a unique, metadata-driven search interface that leverages advanced data management pipelines.
Register for the April 6th webinar: Using NCBI Databases with Tools that Predict Genomic Variant Effects
Thursday, March 24, 2016
In two weeks, NCBI will give a demonstration of some open-source tools that use NCBI databases to predict effects of variants. We will begin with an overview of where to find and download data, particularly VCF and FASTA files, from NCBI, then show you how to use this data in 10 external tools that predict variant functional consequences, including ANNOVAR, PANTHER, SNAP-2, and CBIO MutationMapper.
Date and time: April 6, 2016 1:00 PM EDT
Registration link: https://attendee.gotowebinar.com/register/1347860891564622851
After registering, you will receive a confirmation email with information about attending the webinar. After the live presentation, the webinar will be uploaded to the NCBI YouTube channel. Any related materials will be accessible on the Webinars and Courses page; you can also learn about future webinars on this page.
Register for the April 13th webinar, Submitting Data to NCBI and BioSample
Wednesday, March 23, 2016
In three weeks, NCBI staff will guide you through the process of submitting sequence data to NCBI BioSample. This webinar will show you how to describe samples and sources, and share tips on making submission to BioSample easier.
Date and time: April 13, 2016 1:00 PM EDT
Registration link: https://attendee.gotowebinar.com/register/956885551555521537
After registering, you will receive a confirmation email with information about attending the webinar.
After the live presentation, the webinar will be uploaded to the NCBI YouTube channel. Any related materials will be accessible on the Webinars and Courses page; you can also learn about future webinars on this page.
New NCBI video on YouTube provides strategies to search ClinVar efficiently
Wednesday, March 23, 2016
In the newest video on the NCBI YouTube channel, Search ClinVar with Ease, we share search strategies that will help you search ClinVar, our public archive of reports of relationships between human variations and phenotypes, more efficiently. Learn how to search by gene symbol, variant name and disease, and learn how to browse through variants in a genomic region with Variation Viewer.
Subscribe to the NCBI YouTube channel to receive alerts about new videos ranging from quick tips to full presentations.
RefSeq release 75 is now available
Tuesday, March 15, 2016
RefSeq release 75 is accessible via FTP and through NCBI’s programming utilities. This full release incorporates genomic, transcript and protein data available as of March 7, 2016 and includes 92,936,289 records, 61,034,675 proteins, 14,035,988 RNAs, and sequences from 58,776 organisms.
The release is provided in several directories as a complete dataset and also as divided by logical groupings. More information about release 75 can be found in the release notes. For more information about the RefSeq project, please see the RefSeq homepage.
March 23, 2016: NCBI to offer workshop for advanced SRA and dbGaP users
Monday, March 14, 2016
On March 23 at 12 PM EST, NCBI staff will present a workshop for advanced users of SRA and dbGaP who are interested in using public datasets, and:
- Use and move large genomic datasets,
- Use cloud computing for analyzing genomic datasets,
- Express an interest in doing parallel work on genomic datasets,
- Or are well-versed in RNA-Seq, variant calling, or metagenomics.
The registration link lists the specific topics the workshop will cover. For a more general explanation of NCBI’s genomic resources, please visit NCBI Learn, where we have webinars and factsheets pertaining to dbGaP, SRA, and more.
Search for WGS Sequences using Stand-alone BLAST!
Monday, March 07, 2016
It is now much easier to search WGS (Whole Genome Shotgun) with stand-alone BLAST on your own computer. New tools from the NCBI allow you to BLAST just the WGS projects you are interested in. You can also search a taxonomic subset of WGS (e.g., all human or all bacterial sequences). These new tools for WGS make the existing search mechanism obsolete.
As of August 5, 2016, the current single WGS BLAST database will be retired from the NCBI FTP site and BLAST server. We suggest moving to the new tools as soon as possible.
Read more at ftp://ftp.ncbi.nlm.nih.gov/blast/WGS_TOOLS/README_BLASTWGS.txt.
First of the New Bookshelf NCBI Insights Blog Posts - New Streptococcus pyogenes book
Wednesday, March 02, 2016
The first of a new series of NCBI Insights blog posts highlighting books and documents is available on NCBI’s Bookshelf showcasing a new book: “Streptococcus pyogenes: Basic Biology to Clinical Manifestations”.

Published by the University of Oklahoma Health Sciences Center, this new open-access book provides a comprehensive review of research on the bacterium Streptococcus pyogenes (Group A Streptococcus) which is responsible for diseases such as scarlet fever, pharyngitis, impetigo, cellulitis, necrotizing fasciitis and toxic shock syndrome, as well as the sequelae of rheumatic fever and acute poststreptococcal glomerulonephritis.
“Streptococcus pyogenes: Basic Biology to Clinical Manifestations” is freely available on NCBI’s Bookshelf, at http://www.ncbi.nlm.nih.gov/books/NBK333424/.
NCBI is phasing out sequence GIs - use Accession.Version instead!
Wednesday, March 02, 2016
As of September 2016, the integer sequence identifiers known as "GIs" will no longer be included in the GenBank, GenPept, and FASTA formats supported by NCBI for sequence records. The FASTA header will be further simplified to report only the sequence accession.version and record title for accessions managed by the International Sequence Database Collaboration (INSDC) and NCBI’s Reference Sequence (RefSeq) project. As NCBI makes this transition, we encourage any users who have workflows that depend on GI's to begin planning to use accession.version identifiers instead. After September 2016, any processes solely dependent on GIs will no longer function as expected.
GI numbers have been in use since GenBank release 81.0 (February 1994) as an additional identifier to the accession number to stably refer to a specific version of a sequence record. Version tracking was added to accession numbers in 1997 as an integer suffix that increments with each update to the sequence data within a record. For example, “AC020606.7” indicates that the sequence content of the record has been updated six times since the first release. Thus, sequence versioning information has been provided in a redundant fashion through both the GI and the accession.version. In the past decade, NCBI has continued to receive submissions of new or updated sequences at a rapidly increasing rate. In response to this, we have had to develop new data storage solutions that use accession.version information, rather than GI information, to track updates. Current examples of sequences that lack a GI include unannotated contigs in WGS and TSA projects. This results in a situation where we are conveying version information inconsistently.
Given both the continued increase in the volume of data submissions and the growing inconsistency in record presentation, it is time for us to take the next step and remove the older, redundant GI identifiers and retain a single identifier for sequence versions, the more human-readable accession.version. This change will simplify the process of tracking sequences without any loss of functionality. This change will also simplify scientific communications by promoting use of accession.version as the preferred sequence identifier. Therefore, over the coming months we will no longer assign GI's to an increasing number of new sequences. Sequence records with existing GI's will retain them in some presentation formats, such as ASN.1 and the 5-column feature table format, but the GI value will no longer be displayed in other presentation formats including GenBank flat file and FASTA formats. NCBI services that accept GI's as input will continue to be supported, and NCBI will be adding support for accession.version identifiers to all services that currently do not support them.
This transition to stop assigning and reporting GIs was first described in the Release Notes for GenBank 199.0 in December 2013 and also described in a recent GenBank update. Please see Section 1.4.1 of the current GenBank release notes for background information: ftp://ftp.ncbi.nih.gov/genbank/gbrel.txt
The FASTA display for all sequence records exchanged by the INSDC and for all NCBI RefSeq records will also be changed to report only the accession.version and the record title. This will improve compatibility with other file types provided by NCBI, including GFF3, Gene, and dbSNP download files. This FASTA format change has already been made on data available from the redesigned genomes FTP site based on user requests to have a single consistent sequence identifier for both GFF3 and FASTA formats. See the prior announcement of this change: http://www.ncbi.nlm.nih.gov/news/08-26-2014-new-genomes-FTP-live/ .. At this time, we plan to continue to provide database source information in the FASTA display of sequences from non-INSDC and non-RefSeq sources including: SwissProt, PDB structures, PIR, and patent sequences.
After September 2016, these changes will start to appear on NCBI web views of flat file and FASTA format sequence data, NCBI programming utilities results, and GenBank and RefSeq comprehensive FTP releases.
Example 1: An INSDC nucleotide record - In the sample record below, nucleotide sequence AF123456 was assigned a GI of 6633795, and the protein translated from its coding region feature was assigned a GI of 6633796:

After September 2016, the accession.version will be the sole indicator of the sequence version. The GI value on the VERSION line and the GI /db_xref qualifier for the coding region feature will no longer be visible.
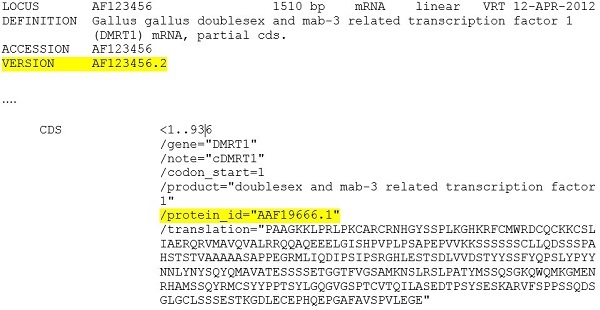
Example 2: A GenPept protein record - The current record display includes the GI in the VERSION lines. Note that the coding region feature for GenPept format has never included the display of GI values.

After September 2016, the VERSION line will not include the GI value:

Example 3: Changes to FASTA format: GI and database source values will be removed from FASTA header - The current FASTA display, in most resources, currently includes GI and database source information (e.g., ‘gb’ for GenBank) delimited with a ‘|’. Downstream analysis tools often require first processing the FASTA header line to simplify the sequence identifier portion to the accession.version or GI. The complex FASTA sequence identifier is highlighted in yellow:

After September 2016, a simple sequence ID will be provided in the FASTA header for nucleotide and protein records

Tree Viewer's Next Update is Available
Wednesday, March 02, 2016
An updated version (v.1.8.0) of the NCBI Tree Viewer, a tool for viewing your own phylogenetic tree data, has been released which has several new features and improvements, as well as some bug fixes.
These include:
- “Link to View” function to create minimized links to Tree Viewer
- Feedback function to inform NCBI developers about issues and improvements
- Mechanism to customize labels
- Better Zoom navigation with adaptive levels and a new button to quickly zoom to the minimal level where node labels become visible
- API zooming functions for embedded views
- New API for creation of custom labels
- Aspect ratio selection to improve the display of radial trees
In addition, several bugs have been fixed.
To see the full list of changes, see the Tree Viewer release notes.
- Specialized database with unique search interface added to Zika virus resource page
- Register for the April 6th webinar: Using NCBI Databases with Tools that Predict Genomic Variant Effects
- Register for the April 13th webinar, Submitting Data to NCBI and BioSample
- New NCBI video on YouTube provides strategies to search ClinVar efficiently
- RefSeq release 75 is now available
- March 23, 2016: NCBI to offer workshop for advanced SRA and dbGaP users
- Search for WGS Sequences using Stand-alone BLAST!
- First of the New Bookshelf NCBI Insights Blog Posts - New Streptococcus pyogenes book
- NCBI is phasing out sequence GIs - use Accession.Version instead!
- Tree Viewer's Next Update is Available
- NCBI News, March 2016 - NCBI NewsNCBI News, March 2016 - NCBI News
Your browsing activity is empty.
Activity recording is turned off.
See more...