Clinical Description
To date, more than 175 individuals from more than 90 families have been identified with prolidase deficiency, as recently reviewed by Spodenkiewicz et al [2020] and Rossignol et al [2021]. The following description of the phenotypic features associated with this condition is based on these reviews.
Note: A minority of individuals (n=5) have remained asymptomatic, despite biochemical or molecular confirmation of prolidase deficiency [Isemura et al 1979, Lemieux et al 1984, Ledoux et al 1996, Mandel et al 2000, Lupi et al 2006]. However, limited long-term information is available on these individuals; the oldest asymptomatic individual was 29 years old at the time of report.
Table 2.
Prolidase Deficiency: Frequency of Select Features
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
| Dysmorphic facial features | 66% | |
| Skin ulcers | 62% | Most commonly affecting lower extremities, particularly feet |
| ID/DD | 58% | IQ ranging from 30 to 90 1 in those who have undergone IQ testing (n=18) |
| Recurrent infections | 48% | Commonly sinopulmonary infections or gastroenteritis |
| Organomegaly | 45% | Mostly splenomegaly, rarely (14%) accompanied by hepatomegaly |
| Skeletal findings | 34% | Incl hand/feet (14%) & other limb (6%) abnormalities, osteopenia (7%), joint hypermobility (7%), or arthritis (5%) |
| Anemia | 30% | |
| Thrombocytopenia | 18% | |
| Chronic pulmonary disease | 12% | Excl asthma (7%) |
| Autoimmune disease | 12% | Incl SLE or SLE-like features, rhupus, 2 autoimmune GI disease or arthritis |
| Hypergammaglobulinemia | 15% | Incl hyper IgG & hyper IgE |
DD = developmental delay; GI = gastrointestinal; ID = intellectual disability; IgG = immunoglobulin G; IgE = immunoglobulin E; SLE = systemic lupus erythematosus
- 1.
The formal medical definition of intellectual disability is an IQ score lower than 70. Individuals with an IQ score equal to or above 70 may still have delays in the acquisition of milestones but would not be considered to have intellectual disability.
- 2.
Rhupus is a condition in which individuals have features of both rheumatoid arthritis and SLE, often in a sequential manner.
Dysmorphic Facial Features
Although not a universal finding, facial features typically described include prominent forehead, widely spaced eyes, proptosis, depressed nasal bridge, prognathism, thin vermilion of the upper lip, and low anterior and posterior hairlines [Falik-Zaccai et al 2010, Besio et al 2015]. See .
Skin
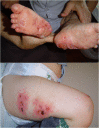
The hallmark of prolidase deficiency is severe, chronic, recalcitrant, and painful skin ulcers (see ). The ulcers are located mainly on the lower extremities, particularly the feet. However, a few individuals have been reported with upper extremity involvement [Sheffield et al 1977, Cantatore et al 1993, El-Darouti 2013] or, rarely, facial involvement [Isik et al 2008].
Skin ulcers can begin as early as age six months or as late as age 30 years. Half of affected individuals have ulcers by age 12 years and around 75% by age 18 years (see also
Genotype-Phenotype Correlations).
Ulceration is recurrent, and individual ulcers can take months to heal.
Typically, no precipitating factors are identified with the appearance of an ulcer, although trauma has been reported as a triggering factor.
A variety of skin findings can precede the appearance of ulcers by many years, including:
Telangiectasias of the face, shoulders, and hands
Scaly, erythematous, maculopapular lesions
Purpuric lesions in the absence of hematologic abnormalities
Premature graying of the hair
Occasional skin findings may also include:
As prolidase deficiency is associated with chronic recalcitrant lower extremity ulcers, an increased risk of squamous cell carcinoma of the skin could be expected, and indeed has been reported in one individual [Fimiani et al 1999].
Developmental Delay (DD) and Intellectual Disability (ID)
DD of variable degree has been described in approximately 60% of individuals with prolidase deficiency [Rossignol et al 2021].
Organomegaly
Splenomegaly is common and variable in severity; in one instance massive splenomegaly (spleen measuring 35 cm) was reported [Nasser et al 2015]. Splenomegaly is sometimes accompanied by hepatomegaly.
Hematologic Manifestations
Anemia is present in around 30% of affected individuals and can be either mild microcytic hypochromic anemia or normocytic normochromic anemia [Dunn et al 2011].
Hemolysis has been described, with reticulocytosis varying from 5.9% [Powell et al 1977] to 8.6% [Powell et al 1974].
Thrombocytopenia is present in around 18% of affected individuals, but recurrent bleeding episodes or easy bruising has been rarely reported in the literature.
Immunologic Manifestations
Recurrent episodes of otitis media, sinusitis, pneumonia, and gastroenteritis are common. A few affected individuals have been reported with hemophagocytic lymphohistiocytosis / macrophage activation syndrome, which required emergent management (see Management) [Chidambaram et al 2021, Rossignol et al 2021].
Immunologic studies may reveal the following (see also Suggestive Findings):
Elevated levels of IgG, IgA, and IgM
Hypocomplementemia
Decreased neutrophil chemotaxis
Increased serum IgE levels, ranging from 20,000 IU/mL to 77,600 IU/mL
Pulmonary Manifestations
Asthma-like chronic reactive airway disease has been described in approximately 7% of affected individuals.
Other forms of chronic pulmonary disease occasionally seen:
Bronchiectasis, chronic lipoid pneumonia, and a cystic fibrosis-like
phenotype with elevated sweat chloride and transepithelial potential difference. One man developed severe progressive restrictive lung disease at age 45 years [
Luder et al 2007].
Progressive lung disease with chest CT findings of mainly cystic lung lesions and ground glass opacity in a Druze woman age 24 years [
Butbul Aviel et al 2012]. She experienced further deterioration in her pulmonary function and secondary pulmonary hypertension and became oxygen dependent.
Pulmonary hypertension requiring supplemental oxygen in an Amish child age six years [
Kelly et al 2010]
Pulmonary fibrosis with pulmonary capillaritis and cystic lung changes in two individuals age five years [
Rayment et al 2019] and 22 years, respectively. The latter individual required oxygen therapy by age 33 years [
Cottin et al 2020].
Autoimmune Disorders and Systemic Lupus Erythematosus-Like Findings
About one fifth of individuals with prolidase deficiency develop an autoimmune disorder. Various manifestations of these disorders may comprise arthritis, nephritis, pericarditis, cytopenias, mouth ulcers, malar rash, and hypocomplementemia (low C3, C4, and CH50 levels).
The most common autoimmune disorder associated with prolidase deficiency is systemic lupus erythematosus (SLE), although not all individuals fulfill the American College of Rheumatology criteria for a diagnosis of SLE.
Reported positive markers include rheumatoid factor, antinuclear antibodies, ANCA, anti-dsDNA, anti-cardiolipin, anti-SSA/Ro, anti-SSB/La, anti-chromatin, anti-RNP, and/or anti-Sm antibodies.
Sometimes, these antibodies have been reported in the absence of SLE-like clinical findings [
Kurien et al 2013].
Other autoimmune conditions may include:
Autoimmune arthritis, including psoriatic arthritis or juvenile
idiopathic arthritis
Vasculitis, including central nervous system vasculitis
Rhupus (a condition in which individuals have features of both rheumatoid arthritis and SLE, often in a sequential manner)
Coombs-positive hemolytic anemia
Crohn-like disease
Bone/Limb Manifestations
Among 12 affected individuals reported by Besio et al [2015], short stature was described in seven, osteopenia in six, and genu valgum in four. Joint laxity has been described in more than a dozen affected individuals. Some limb deformities have been reported to be secondary to complications of severe ulceration, particularly in the lower limbs. Talipes equinovarus was noted in early reported cases and may be a secondary finding. Digital clubbing, in the presence or absence of pulmonary abnormalities, has been reported in up to 7% of affected individuals [Luder et al 2007, Kelly et al 2010].
Other primary bone/limb findings (reported in a few cases each) include:
Other Manifestations
Ocular abnormalities have been found in approximately 15% of affected individuals and include varied phenomena including optic and chorioretinal atrophy in two individuals [Ogata et al 1981] and keratitis [Ogata et al 1981, Freij et al 1984, Andry et al 1992].
Dental abnormalities were present in more than 20 affected individuals. Dental findings have included multiple caries with or without enamel hypoplasia, misaligned teeth, and hypodontia.
Diarrhea has been reported in 19 affected individuals, even in the absence of Crohn-like inflammatory bowel disease (reported in only 5 individuals).
Both poor weight gain (14%) and short stature (12%) as well as overweight/obesity (9%) have been reported.
Prognosis
The severity of prolidase deficiency is quite variable: in some individuals skin ulcerations lead to amputation of one or all toes, whereas others remain entirely asymptomatic [Isemura et al 1979, Hechtman 2001, Lupi et al 2006]. Most individuals will have developed some symptoms (albeit sometimes mild) of prolidase deficiency by age four years and 90% by age 14 years.
In many instances, however, individuals with prolidase deficiency experience severe morbidity and sometimes early death, usually as a result of infection or respiratory failure. The youngest reported death was at age three months [Shrinath et al 1997]. A review by Rossignol et al [2021] estimated a survival of 90% by age 20 years and 82% by age 40 years.
Prevalence
Approximately 175 affected individuals have been reported in the literature; however, prolidase deficiency likely remains underdiagnosed as a result of underrecognition by physicians.
The Québec Newborn Urine Screening Program (Programme québécois de dépistage neonatal urinaire, PQDNU) identified two affected infants out of 2,469,929 screened between 1973 and 2006, for an incidence of 1:1,235,000 [Renaud & Dagenais 2009]. Thus, a few thousand cases would be predicted to exist worldwide, as opposed to only about 175 cases reported to date.
Prolidase deficiency has been diagnosed throughout the world [Lupi et al 2008].
A founder variant has been described in the Geauga County Amish settlement in Ohio [Wang et al 2006], as well as in the Druze (carrier frequency 1:21) and Arab Muslim populations in northern Israel [Falik-Zaccai et al 2008] (see Molecular Genetics).